|
|
|
 |
|
|
|
Molecular Collisions Laboratory
Brian Stewart
Wesleyan University
Department of Physics
bstewart@wesleyan.edu
|
|
|
|
The research in the Molecular Collisions Group is aimed at understanding the different kinds of few-body dynanics that can occur in the context of atom-molecule collisions. It is perhaps surprising that even now there are qualitatively new kinds of dynamics to be discovered in this simple physical context. |
|
|
|
 |
|
|
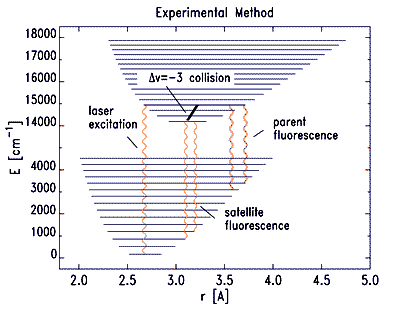
At present, our experimental system is Li2 - Ne, a non-reactive collisional system in which we measure level-resolved energy transfer rate coefficients and cross sections. The collisions occur on a single excited-state potential surface; population is transferred from the ground electronic state to a single rovibronic level by means of a single-frequency dye or diode laser. The ensuing emission is dispersed, and the intensities of the resulting spectral lines are used as measures of the excited-state population densities. By varying the Ne density, we can obtain the level-to-level rate coefficients from solution of the coupled rate equations for the various collision and photophysical processes that occur in the excited state. |
|
|
|
 |
|
|
| The experiments take place in a heated cell; consequently, there is a thermal distribution of collision speeds. To extract the speed-dependent cross section, it is necessary to vary the freqency of the laser, thus selecting different velocity groups from the Doppler profile of the excitation. Through a deconvolution, the detuning-dependent rate coefficients may be converted into speed-dependent cross sections. |
|
|
|
 |
|
|
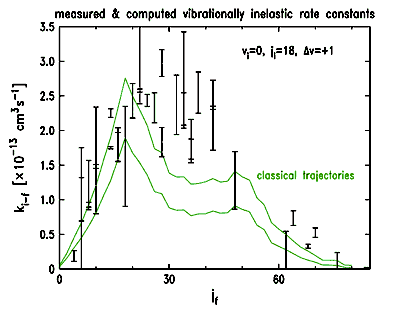
To complete these studies, we model the collisions using classical trajectories and full quantum mechanical calculations. Typically, the classical trajectory calculations on an ab initio potential surface agree very well with experiment.
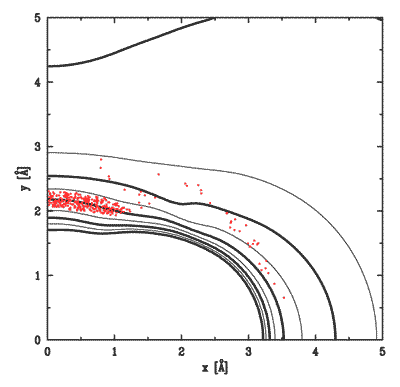
As an example of unexpected collision dynamics, we cite the surprising mode of vibrational excitation of initially unexcited Li_2 molecules. Here our classical calculations serve to guide us in choosing experimental conditions. The accompanying figure shows equipotentials of the Li_2 - Ne potential energy surface. Superposed are dots, placed at the classical turning point of each collision that successfully excited the molecular vibration. It is evident that equatorial impacts, rather than polar impacts, are most effective in exciting the vibration. Yet the collisional coupling at the equator is far weaker than that at the poles. The reason for this turns out to be a strictly dynamical effect -- near-polar impacts lead early in the collision to significant rotational excitation, which reduces the energy available for vibrational excitation. Rotational excitation effectively quenches vibrational excitation.
|
|
|
|
Depicted above are equipotentials of the Li2 - Ne potential surface. Superposed are the classical turning points of trajectories that excited the molecular vibration. Equatorial impacts are preferred over polar impacts. |
|
|
 |
|
|
| Equatorial impacts, unlike those near the molecular pole, do not exert a torque on the molecule. Hence there is no rotational excitation to remove energy from the collision and "quench" the vibrational excitation. Our data so far are consistent with this phenomenon -- note the steep rise to a maximum at a jf value near ji. (There are no adjustable parameters in either the data or the computation.) Yet there is not perfect agreement, either. We are currently calculating quantum mechanical rate coefficients on the ab initio potential surface to aid us in a more detailed analysis of the differences and similarities between the experimental and computed rate constants. |
|
|
|
 |
|
|
|
Publications:
A. Billeb and B. Stewart, Chem. Phys. Lett. 247, p. 433 (1995)
Y. Gao, P. S. Gorgone, S. Davis, E. K. McCall, and B. Stewart, J. Chem. Phys. 104, p. 1415 (1996).
B. Stewart, P. D. Magill, and D. E. Pritchard, J. Phys. Chem. A 104, p. 10565 (2000).
|
|
|
|
Group members Ethan Butler, Paula Matei, Steve Coppage, and Nasim Khoshkhou in a moment of group hilarity. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|